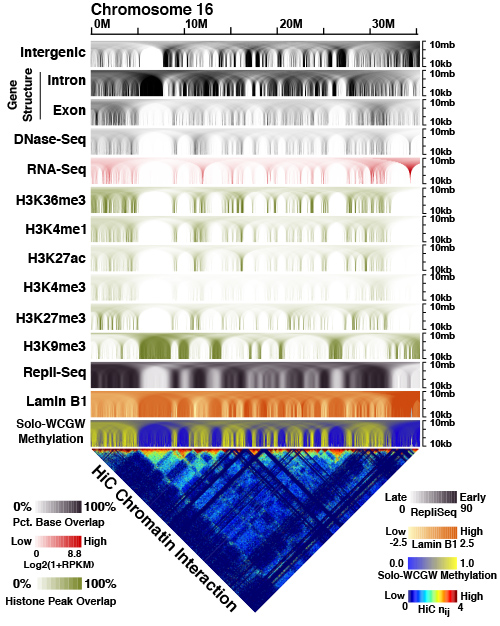
Global methylation loss patterns in aging and cancer
Most CpG dinucleotides are methylated at birth, but replication errors during life and especially in cancer lead to global hypomethylation of the genome. We are interested in how this loss of methylation allows expression of normally silenced genes and repetitive elements, and how these events can promote cancer or evoke an immune response. We provided the first comprehensive atlas of these methylation regions (Zhou, Nature Genetics 2018).
We uncovered how different CpGs lose methylation at different rates, and developed a deep learning model to predict these rates based on sequence context alone (Zheng, bioRxiv 2022), which allowed us to identify a set of cancer-specific changes and develop a computational tool (MMSeekR) to identify differences between different cancer types (Zheng, Genome Biology 2023). We are interested in how these changes predict response to immune therapies.
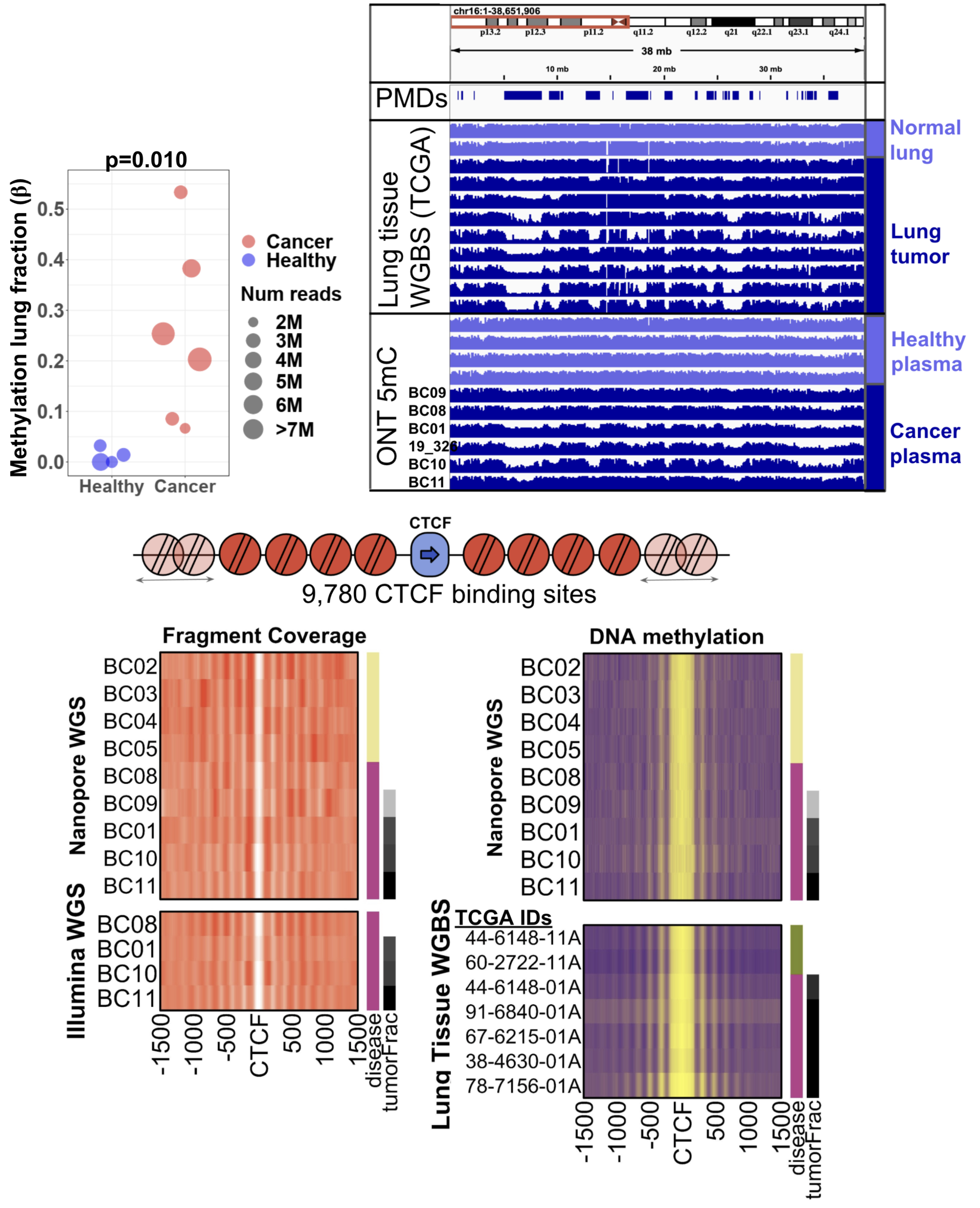
Characterizing tumor DNA in circulation using Oxford Nanopore sequencing
Cancer cells shed genomic DNA into circulation, and “liquid biopsies” are increasingly common to detect and characterize a tumor without an invasive biopsy of the tumor itself. We use Oxford Nanopore whole-genome sequencing, which allows us to characterize not only the DNA sequence but the DNA methylation patterns of the tumor DNA.
We showed that multiple complementary genomic features could be used to identify circulating tumor DNA (Katsman, Genome Biology 2022), which included characterization of methylation loss patterns. One of the major benefits of Nanopore sequencing is the ability to identify specific cell types using specific DNA methylation markers. We developed a leading method for deconvolution of cell types called CelFiE-ISH (Unterman, Genome Biology 2024), and are interested in advancing these techniques even further.
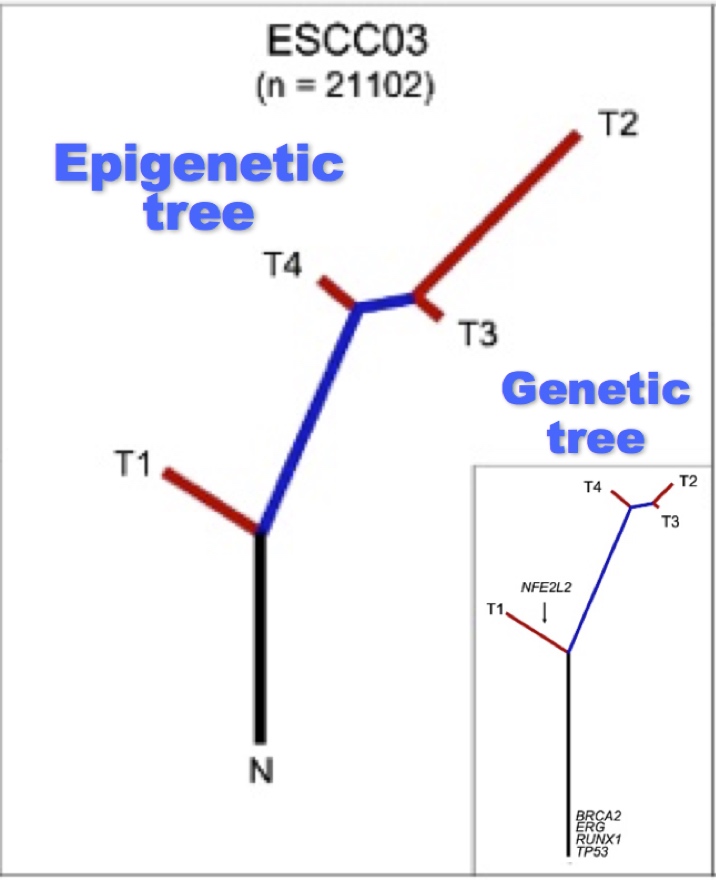
Understanding cancer evolution through epigenetic intra-tumour heterogeneity
Tumors are not monolithic, and often have multiple subclones with different alterations. We are interested in how methylation changes allow different subclones to adapt to different functional constraints, especially immune evasion. We surveyed genomic methylation changes across subclones, and found that DNA methylation changes followed a similar evolutionary path as the genome in esophageal cancer (Hao et al., Nat. Genetics 2016), but were highly diverged in liver cancer (Lin et al., Cancer Res.2017). By re-analyzing global methylation patterns in single-cell methylation datasets, we found that global methylation loss can be dramatically different in different subclones (Zheng, bioRxiv 2022), suggesting that immunogenic responses to global hypomethylation may by subclone specific.
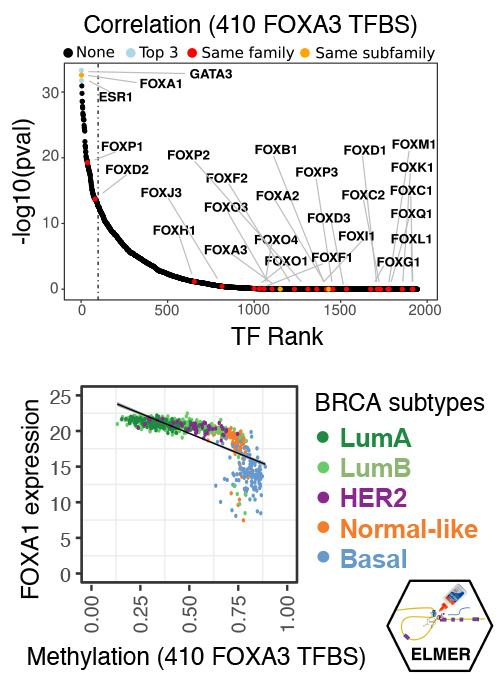
Reconstructing altered transcription factor networks from chromatin changes at cis-regulatory elements
Our lab is interested in the reconstruction of cancer gene regulatory networks from DNA methylation and other chromatin changes that occur at transcription factor binding sites (TFBSs). For instance, we collaborated with Peter Jones’ lab to develop the multi-omic NOMe-seq technique for determining the differing roles of DNA methylation and chromatin accessibility at TFBSs (Kelly et al., Genome Res. 2010), and developed a computational approach Bis-SNP to deconvolute methylation and SNP information from bisulfite sequencing to identify allele-specific TFBSs (Liu et al., Genome Biol. 2012). We developed a Bioconductor R package, ELMER, that integrates DNA methylation information at TFBSs with TF expression levels to reconstruct gene regulatory networks (Yao et al., Genome Biol. 2015; Silva et al., Bioinformatics 2019).
